Single Nucleotide Polymorphisms of Human STING
2018-02-28
How amino acid variations affect innate immune response to foreign DNA
Understanding how the innate immune system senses foreign DNA generated by certain viruses, bacteria, and other non-self nucleic acids is significant to developing potential treatments for microbial-related disease, including viral-associated cancers and autoimmune disorders. The identification of the endoplasmic reticulum-associated protein referred to as STING (stimulator of interferon genes) has been pivotal for revealing how type 1 interferon (IFN) production is triggered in response to intracellular DNA and DNA from pathogens. Studies comparing cyclic dinucleotide (CDN) binding and STING activation identified significant single nucleotide polymorphisms of human STING.
The five human haplotypes are known as WT (R232), REF (R232H), HAQ (R71H, G230A, R293Q), AQ (G230A, R293Q), and Q (R293Q).1,2 Each has evolved differently to distinguish conventional bacterial CDNs (i.e., 3ʹ3ʹ-cGAMP containing G(3ʹ5ʹ)pA(3ʹ5ʹ) linkages, c-di-GMP, and c-di-AMP) from noncanonical metazoan CDNs (i.e., 2ʹ3ʹ-cGAMP containing G(2ʹ5ʹ)-pA(3ʹ5ʹ) phosphodiester linkages).2 For instance, the R232H allele, which occurs in 14% of the population, responds to 2ʹ3ʹ-cGAMP, but responds weakly to bacterial CDNs. In contrast, STING-HAQ, which is found in 20.4% of the population, is a loss-of-function variant that responds weakly to both metazoan and bacterial cGAMPs.1,3 Furthermore, STING-Q, which occurs in 1.5% of the population, dramatically decreases ability to respond to all bacterial ligands. Interestingly, the biased distribution of haplotypes suggests specific selection for certain protein isoforms in geographically separated populations.1 STING-AQ and STING-Q isoforms occur predominantly within the African subpopulation, while HAQ is much higher in Asian and Hispanic American subpopulations. The WT and R232H variants occur across all populations. While all STING haplotypes possess some ability to recognize metazoan CDNs, this suggests potential geographical differences in the ability of subpopulations to respond to bacterial CDNs.
Researchers have also begun to introduce point mutations into STING to further understand the amino acids important for CDN binding and/or IFN induction. What is known so far is that STING binds to CDNs through its C-terminal, cytoplasmic domain. Two STING molecules bind one cyclic-di-GMP, implying that a STING dimer shares one CDN binding site (Figure 1).4
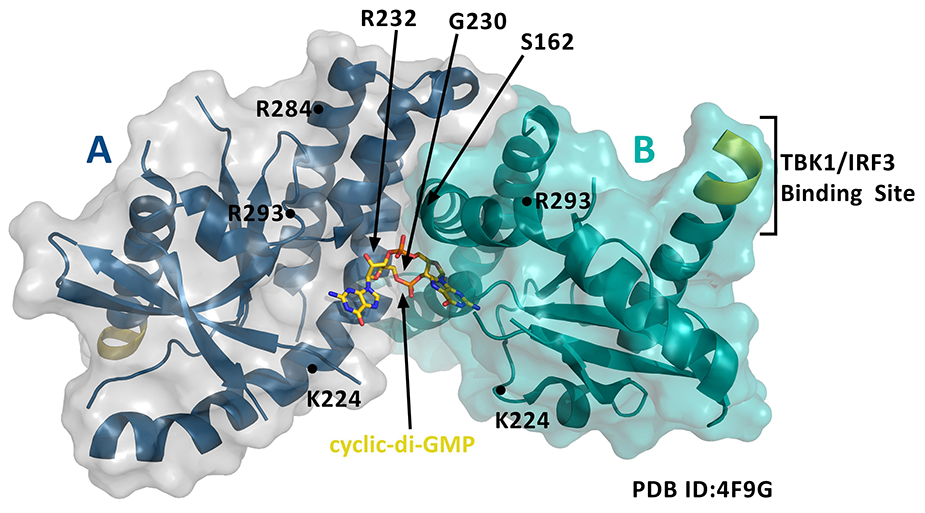
Figure 1. STING dimer (A and B are both aa 139-379) bound to cyclic-di-GMP and notated with key amino acid modifications of Cayman's STING variants.
Residues 153-177 form part of helix α1 and are involved in the dimerization of STING and the binding of CDNs (Figure 2).5 Cayman offers several human recombinant variants of the STING protein expressed in E. coli, some of which occur as natural haplotypes and others of which introduce point mutations within the CDN binding domain. Because N-terminal, transmembrane domain deletion truncations of full-length STING can be expressed as highly soluble proteins,6 Cayman’s STING variants include the key, soluble residues of the C-terminal domain. Known differences in how these variants respond to CDNs and/or affect the downstream IFN response are noted below. Use the sequence map in Figure 2 to keep track of the amino acid substitutions in Cayman’s STING variants.
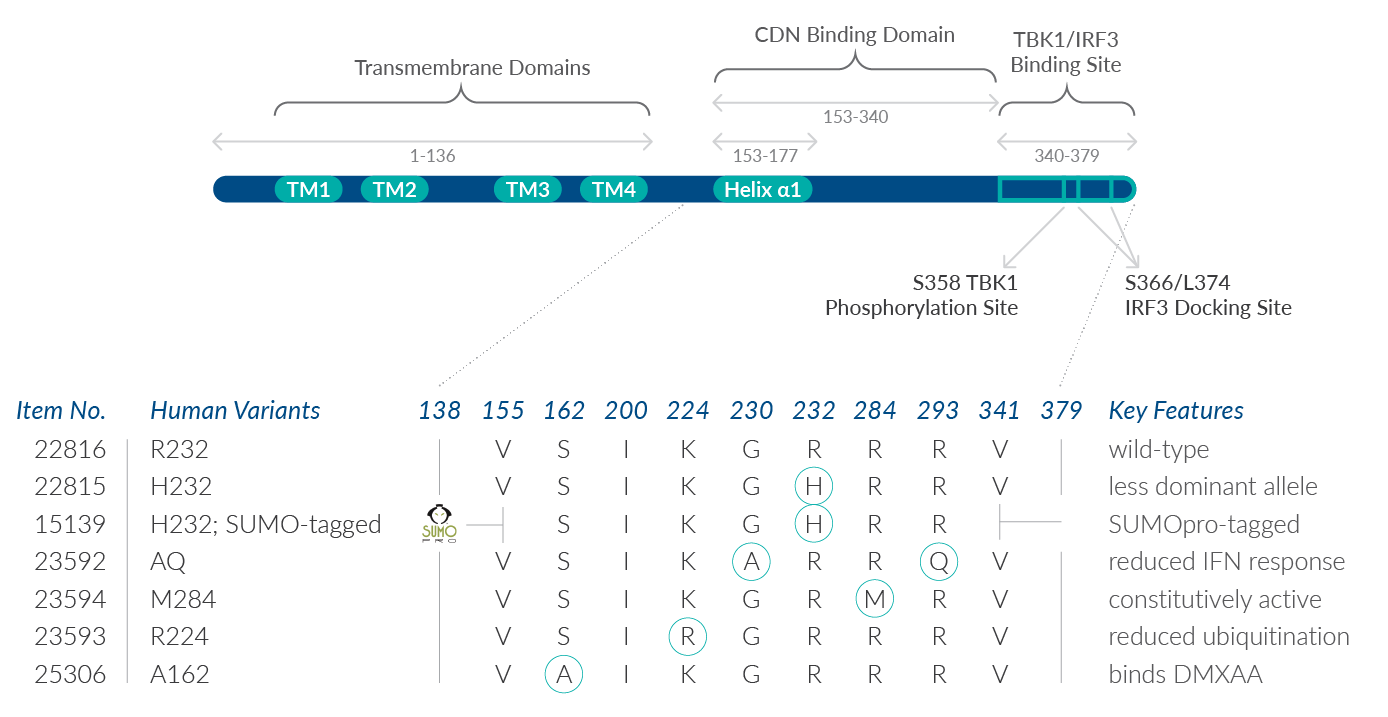
STING R232 variant (human recombinant) contains amino acids 138-379 with an arginine at position 232. This is the most prevalent human STING isoform, occurring naturally in 60% of the human population, and thus is considered as “wild type.”1 R232 is located in the loop structure that is predicted to form the c-di-GMP binding pocket of STING. In addition to c-di-GMP and c-di-AMP, it can bind both 2'3'- and 3'3'-cGAMPs with preferential activation via metazoan (2’5’-linkage-containing) cGAMP isomers.1,7
STING H232 variant (human recombinant) contains amino acids 138-379 with a histidine at position 232 in place of the arginine. This R232àH variation is found in 13.7% of the human population.1 H232 binds metazoan 2’3’-cGAMP but demonstrates a partially reduced IFN response to bacterial c-di-GMP and a complete loss of IFN response to c-di-AMP and 3’3’-cGAMP.1,7 Many structural studies of human STING proteins and early characterizations of its activity have used the H232 allele.
STING H232 variant; SUMO-tagged (human recombinant) contains amino acids 155-341 of the H232 variant and a removable N-terminal SUMOpro tag. With both N- and C-terminal truncations, this variation presents only the central CDN binding domain, eliminating four transmembrane domains (aa 1-136) and the tail-end TBK1/IRF3 binding sites (aa 340-379). Note that a SUMO tag may interfere with certain studies. SUMOylation of cGAS (which functions upstream of STING) has recently been shown to dampen cGAS activation and impair IRF3-responsive gene expression.8
STING AQ variant (human recombinant) contains amino acids 138-379 of the wild-type variant with an alanine at position 230 instead of a glycine and a glutamine at position 293 instead of an arginine. These co-segregating substitutions occur in 5.2% of the human population and demonstrate a partially reduced IFN response to bacterial ligands (30-40% reduction compared to wild type).1 Whereas the R293àQ substitution alone is severely defective in response to bacterial CDNs, the G230àA substitution (which alters the conformation of the lid region that clamps on to c-di-GMP) is thought to help maintain some ability of this variant to respond to bacterial CDNs.1 Key amino acid residues near 293 are speculated to be important for IFN stimulation.9
STING M284 variant (human recombinant) contains amino acids 138-379 of the wild-type variant with a methionine at position 284 instead of an arginine. This mutant is associated with constitutive activation of downstream signaling. That is, R284àM has been shown to increase the propensity of STING to dimerize and associate with the kinase TBK1, enhancing the ability of STING to activate IRF3 and NF-κB and induce a type I IFN response.10 Since this mutation at 284 occurs outside of the 153-177 region that is key for dimerization, R284M is predicted to promote binding of a cellular factor that stabilizes STING dimerization.10 Conversely, the R284M could potentially inhibit binding of a cellular factor that impairs STING dimerization.
STING R224 variant (human recombinant) contains amino acids 138-379 of the wild-type variant with an arginine at position 224 instead of a lysine. Ubiquitination of STING on K224 by the mitochondrial E3 ubiquitin protein ligase 1 is essential for efficient cytosolic DNA-mediated signaling.11 By reducing the ubiquitination of STING, the K224àR substitution interrupts optimal STING trafficking. K224R has been shown to inhibit TBK1-mediated IRF3 activation but not NF-κB activation.11
STING A162 variant (human recombinant) contains amino acids 138-379 of the wild-type variant with an alanine substituted for serine at position 162. This point mutation is located in the CDN binding site and allows human STING to bind to DMXAA, a compound previously known only to bind mouse STING.12,13 When bound to DMXAA, A162 adopts the closed conformation, similar to the conformation it has when bound to the second messenger 2’3’-cGAMP, and activates TBK/IRF3 signaling.12
Immunotherapeutic Potential
The importance of STING in facilitating innate immune responses following infection with DNA viruses or microbes makes this pathway a key target for immunotherapeutics. The STING variants that Cayman offers can be used to better understand the key amino acids involved in recognizing noncanonical versus bacterial CDNs and triggering a specific IFN response.
Download the Innate Immune Signaling Currents to learn more about the products Cayman offers for this area of research.
BULK & CUSTOM ORDERS AVAILABLE
We can scale up production, synthesize custom STING mutants, remove purification tags, and help advise on which variant is the best fit for your research. Email us at sales@caymanchem.com for more information.
References
1. Yi, G., Brendel, V.P., Shu, C., et al. PLoS One8(10), e77846 (2013).
2. Diner, E.J., Burdette, D.L., Wilson, S.C., et al. Cell Rep.3(5), 1355-1361 (2013).
3. Patel, S., Blaauboer, S.M., Tucker, H.R., et al. J. Immunol. 198, 776-787 (2017).
4. Shu, C., Yi, G., Watts, T., et al. Nat. Struct. Mol. Biol.19(7), 722-724 (2012).
5. Huang, Y.-H., Liu, X.-Y., Du, X.-X., et al. Nat. Struct. Mol. Biol.19(7), 728-731 (2012).
6. Ouyang, S., Song, X., Wang, Y., et al. Immunity36(6), 1073-1086 (2012).
7. Gao, P., Ascano, M., Zillinger, T., et al. Cell 154(4), 748-762 (2013).
8. Cui, Y., Yu, H., Zheng, X., et al. PLoS Pathog.13(1), e1006156 (2017).
9. Jin, L., Xu, L.-G., Yang, I.V., et al. Genes Immun. 12(4), 263-269 (2011).
10. Tang, E.D. and Wang, C.-Y. PLoS One10(3), e0120090 (2015).
11. Ni, G., Konno, H., and Barber, G.N. Sci. Immunol.2(11), eaah7119 (2017).
12. Gao, P., Ascano, M., Zillinger, T., et al. Cell 154(4), 748-762 (2013).
13. Conlon, J., Burdette, D.L., Sharma, S., et al. J. Immunol.190(10), 5216-5225 (2013).
